05 Aug 2013
Part of an introductory course on whole-genome shotgun metagenomic sequence analysis, I thought it might be useful to reproduce here.
I am working on building a regularly updated PAUDA and LAST index of microbial-RefSeq protein sequences that might be useful for those who would like to save some time, or don't have enough memory to build these indices.
Taxonomic assignments
Reference databases used for taxonomy
|
Source
|
Database
|
Notes
|
Link
|
|
NCBI
|
Non-redundant proteins (nr)
|
Pre-built BLAST database, use fastacmd to extract sequences
|
ftp://ftp.ncbi.nlm.nih.gov/blast/db/
|
|
NCBI
|
Non-redundant nucleotides (nt)
|
Pre-built BLAST database
|
ftp://ftp.ncbi.nlm.nih.gov/blast/db/
|
|
NCBI
|
Microbial RefSeq genomes
|
|
ftp://ftp.ncbi.nlm.nih.gov/refseq/release/microbial/
|
|
NCBI
|
Microbial RefSeq proteins
|
*.faa files
|
ftp://ftp.ncbi.nlm.nih.gov/refseq/release/microbial/
|
|
NCBI
|
RefSeq genomes
|
Huge
|
ftp://ftp.ncbi.nlm.nih.gov/refseq/release/complete/
|
|
NCBI
|
RefSeq proteins
|
Huge
|
ftp://ftp.ncbi.nlm.nih.gov/refseq/release/complete/
|
|
HMPDACC
|
HMPREFG
|
Human body-site specific
|
http://www.hmpdacc.org/HMREFG/
|
|
Metaphlan
|
Metaphlan lineage-specific
|
Tiny, only considers lineage-specific sequences, pre-built for Bowtie2 or BLAST |
http://huttenhower.sph.harvard.edu/metaphlan |
For further databases and more information, see the useful page from the CAMERA project: http://camera.calit2.net/camdata.shtm
Comparison of software for reference-based taxonomic classification
|
Programme
|
Input
|
Space
|
Sensitivity
|
Speed
|
|
BLASTN
|
Contigs or reads
|
nuc
|
-
|
Slow
|
|
BLASTX
|
Contigs or reads
|
prot
|
++
|
Slow
|
|
BLAT
|
Contigs or reads
|
nuc or prot
|
-/+
|
Medium
|
|
PAUDA
|
Reads
|
ambiguous prot
|
+
|
Fast
|
|
LAST
|
Contigs or reads
|
nuc/prot
|
-/+
|
Fast
|
|
Metaphlan
|
Reads
|
nuc
|
?
|
V.fast
|
|
BWA
|
Reads
|
nuc
|
-
|
V.fast
|
|
Bowtie2
|
Reads
|
nuc
|
-
|
V.fast
|
|
Rapsearch2
|
Reads
|
nuc/prot
|
+
|
Medium
|
Others: BLASTZ, mpiBLAST (@yokofakun), USEARCH (@guyleonard)
Rapsearch2
Download NR <ftp://ftp.ncbi.nih.gov/blast/db/FASTA/nr.gz>.
Rapsearch2 takes FASTA as input, so convert using:
seqtk seq -A <MYREADS>.fastq > <MYREADS>.fasta
gunzip nr.gz
prerapsearch -d nr.rap -n nr
rapsearch -q <MYREADS>.fasta -d nr.rap -z 16 -o <MYREADS>.rap -b 50 -v 50 -a t
Import to MEGAN via the command-line
echo "load taxGIFile='/mnt/phatso/nick/rapsearchdb/gi_taxid_prot.bin';" > "$fn".cmd
echo "load keggGIFile='/mnt/phatso/nick/rapsearchdb/gi2kegg.map';" >> "$fn".cmd
echo "import blastFile=$fn.rap.aln meganFile=$fn.rap.rma maxMatches=100 minScore=50.0 maxExpected=1.0 topPercent=10 minSupport=5 minComplexity=0.44 useMinimalCoverageHeuristic=false useSeed=false useCOG=false useKegg=true paired=false useIdentityFilter=false textStoragePolicy=0 blastFormat=RapSearch mapping='Taxonomy:GI_MAP=true,KEGG:GI_MAP=true';" >> "$fn".cmd
echo "quit;" >> "$fn".cmd
xvfb-run -a MEGAN5/MEGAN -g -c "$fn".cmd
How to fetch Microbial Protein Sequences
The easiest and fastest way is via the command-line, using the rsync protocol which is supported by NCBI. The advantage of rsync here is that it will only sync updates, so it is tolerant of breaks in connection, and you can run it regularly and it will only fetch files that have changed.
rsync -avz rsync://ftp.ncbi.nlm.nih.gov/refseq/release/microbial/ \
--include "*/" --include "*.faa.gz" --exclude "*" .
This command will download all files matching the pattern '*.faa.gz', i.e. microbial protein sequences.
rsync -avz rsync://ftp.ncbi.nlm.nih.gov/refseq/release/microbial/ \
--include "*/" --include "*.genomic.gz" --exclude "*" .
Now we need to concatenate them all together:
zcat *.gz > microbial_refseq.faa
Taxonomic assignments with LAST
Creating the LAST database
Before we can search our reads, we need to build the database.
$ bin/last-291/src/lastdb microbial.lastdb <(zcat refs/microbial*)
This is an example of redirection: we are redirecting the output of the command 'zcat refs/microbial*' as the FASTA input. This saves us having to decompress and concatenate the file.
lastdb: that's some funny-lookin DNA
Woops! We need to provide -p option to tell LAST we are indexing a protein database.
$ bin/last-291/src/lastdb -p microbial.lastdb <(zcat refs/microbial*)
This process takes an hour or two on a fast server, and consumes quite a lot of memory.
Assigning reads with LAST
LAST will take FASTQ files, but only for nucleotide databases. We are going to use the six-frame translated mode, as indicated by the -F15 option. In this mode, LAST requires FASTA output!
Here's the l33t way to do it:
time lastal -F15 microbial.last \
<(seqtk seq -A datasets/ecoli/subsample/1122-H_1.fastq) \
>1122_H_1.maf
real 3m25.313s
user 3m15.316s
sys 0m10.028s
This is the same as:
seqtk seq -A datasets/ecoli/subsample/1122-H_1.fastq > 1122_H_1.fasta
lastal -F15 microbial.last 1122_H_1.fasta > 1122_H_1.maf
But you save having to create an intermediate FASTA file.
Questions: How long did it take to assign 100,000 reads with LAST?
Convert LAST results to BLAST format
LAST outputs MAF format which cannot be read by MEGAN without conversion, which we will use later. Additionally, the MAF to BLAST converter supplied with LAST does not produce exactly the right format, please use my modified version instead. The MAF files first need to be sorted by the read identifier (-n2), and then converted, e.g.
maf-sort.sh -n2 1122-H.maf | maf-convert.py blast 1122-H.maf > 1122-H.blast.txt
Running Metaphlan
Metaphlan can use either BLAST or Bowtie2 for assignments. Bowtie2 is significantly faster, so we will use that. It should be already on your path.
metaphlan.py <fastq_file> \
--bt2_ps sensitive-local \
--input_type multifastq \
--bowtie2db software/metaphlan/bowtie2db/mpa \
-t rel_ab \
-o <relative_abundances_file>
metaphlan.py 2535b-H-STEC_1.fastq \
--bt2_ps sensitive-local --input_type multifastq \
--bowtie2db software/metaphlan/bowtie2db/mpa \
-t rel_ab \
-o 2535b.relativeabundances.txt
05 Aug 2013
If you are into sequencing, genomics and bioinformatics, here are a couple of must-attend dates for your diary!
This September, kicking off the second "conference season" of the year, is UK Genome Science 2013. This is one of the largest meet-ups of genomics people in the UK. I like to say it is our version of AGBT, minus the sun, tropical island, palm-trees, pelicans, free booze, all-night parties, exclusive suites with hot-tubs, Jay Flatley playing beer pong. Instead, Nottingham boasts some of the oldest pubs in the land (and thus world) and a willing group of volunteers to show you around them. Excitingly, Clive Brown, CTO of Oxford Nanopore will be giving the keynote, and hopefully updating us on the status of their eagerly anticipated single-molecule sequencer. There are sessions on bioinformatics, metagenomics, clinical application of HTS, pathogens, technologies and AgriFoods. Registration closes this Friday, 9th August, so please register soon!
The fun continues straight after this with another installation of our balti and bioinformatics meetings, this time rebranded for the summer as cream teas and bioinformatics, and hosted by the good folk at the University of Exeter. Come for an informal discussion of the bioinformatics of sequencing, including an opportunity to ask questions of our speakers about your particular bioinformatics problems.
Hope to see you at one or both of these meetings!
24 Jul 2013
The Balti and Bioinformatics juggernaut rolls on, and this September we are going on our summer holidays, to Exeter! So, the next Balti and Bioinformatics is being rebranded as Cream Teas and Bioinformatics!
[caption id="attachment_1731" align="alignright" width="300"] Cream Tea done the correct Devonshire way, with jam on top![/caption]
Cream Tea done the correct Devonshire way, with jam on top![/caption]
We have the excellent Konrad Paszkiewicz, Thomas Laver and James Harrison to thank for hosting the next meeting in this (loosely) themed series.
The meeting will be on the afternoon of Thursday, September 5th, after the UK Genome Sciences meeting in Nottingham which I also urge you to attend!
Registration is free at this link!
Venue is the Institute of Arab and Islamic Studies, lecture theaters 1&2. Building 16 on the map.
http://www.exeter.ac.uk/visit/directions/streathammap/</wbr>
General travel/parking info for Exeter's Streatham campus:
http://www.exeter.ac.uk/visit/directions/streatham/</wbr>
http://www.exeter.ac.uk/visit/directions/carparks/</wbr>
We’ll also be heading out for an evening meal for those who want to stay later. It’ll have a Moroccan theme!
Exeter is not as far away as you may think - 2 1/2 hours from both Birmingham and London by train.
Here's the draft itinerary, subject to amendment:
12.30-1.00: Arrival & coffee
1.00-1.15: Introduction
1.15-1.45: Simon Andrews (Babraham Institute): “SeqMonk – NGS analysis on your desktop”
1.45-2.15: Felix Krueger (Babraham Institute): “Measuring DNA methylation – What could possibly go wrong?”
2.15-2.45: David Studholme (University of Exeter): “Applying high-throughput genome sequencing to plant pathology ”
2.45-3.15: Break for cream teas & coffee
3.15-3.45: Guy Leonard (Natural History Museum): “Genome-scale comparative analysis of gene fusions, gene fissions, and the fungal tree of life”
3.45-4.15: Yannick Wurm (Queen Mary - University of London): “A social chromosome and some bioinformatics challenges in ants”
4.15-4.45: James Abbott (Imperial College London ): “BugBuilder - Hand Free Microbial Genome Assembly"
4.45-5.00: Bioinformatics clinic or/& general discussion
5.00-: Buffet in the sun!
Note, the original version of this post mistakenly said Wednesday. It is on Thursday.
18 Jul 2013
I asked a question on Twitter yesterday:
What is the correct way to handle a request to “help me learn bioinformatics” from a non-computer-literate person?
This is a frequent request I encounter, and although I have various stock answers, I was curious to find out what you guys would say. Further, I wanted a resource with jumping off links, which I hope this blog post can serve as.
And, as with many times before, I was blown away by the use of Twitter for the diversity and quality of opinion and information generated from such a seemingly innocuous question. I know I am lucky in having >3,500 eager followers hanging on my every question, but still ;)
Luis Pedro Coelho initially took issue with the question and said it is important to define what a user really wants, as "learning bioinformatics" "is not a goal per se". Is the user driven by intellectual curiosity, do they want to learn to code, or simply develop enough skills to "analyse my RNA-Seq data?". Russell Neches takes the extreme view which is that bioinformatics "is a fiction. Biologists just use computers for certain things". Manoj Semanta likes to stratify bioinformatics into five layers of increasing complexity, with using web interfaces and running command-line programmes being the easiest and developing new algorithms being the hardest..
Some suggested learning R or Python and pointed to helpful online tutorials, such as http://learnpythonthehardway.org/ [Phil Ashton]. Another useful resource was the excellent Software Carpentry website which is aimed specifically at scientists wishing to learn best practices, for example the use of version control and Makefiles for reproducible research (software-carpentry.org) [suggested by Rob Davey, Deanna Church]. Software Carpentry plan to run more bootcamps for complete novices in the coming year, so keep an eye on their website.
Casey Bergman suggested using Galaxy, a web-based bioinformatics engine particularly heavily used for NGS/genomics analysis (www.galaxy-project.org). Although Rob Davey qualified this advice pointing out that "using a workflow UI and being a bioinformatician may not be the same thing. Chris Cole agrees, "Galaxy != bioinformatics. It's a great and powerful system, tho."
The always opinionated Mick Watson suggested more of a 'sink or swim' approach, specifically to go away and install Ubuntu on a PC or laptop, because "because to "learn bioinformatics" you need commitment and time and effort". Mick also pointed to some online resources hosted at ARK Genomics which expanded on this idea of bioinformatics being tough, but worth investing time in learning (http://www.ark-genomics.org/events-online-training/eu-training-course).
The theme of "learning by doing" is probably the one that I suggest most to people, also suggested by Aylwyn Scally. I tell people that learning programming through reading a book and doing simple exercises is demotivating if you don't have a problem in mind. So pick a problem that you think can be solved with scripting or bioinformatics tools, perhaps a biological question, and attempt to do it "by all means necessary". Being driven by a goal will help you keep motivated. Mario Caccamo says "Learning by doing is not ideal but that's the reality".
Some suggested attempting some simple tasks. One suggestion was to take data from a laboratory evolution paper (e.g. http://www.ncbi.nlm.nih.gov/pubmed/21940899) and try to reproduce it, in this case by detecting a small set of mutations [@contaminatedscience]. Another was to use the intriguing ROSALIND platform to attempt bioinformatics problem solving (http://rosalind.info/problems/locations/) [Robert Lanfae, Adam Kiezun].
Bastien Chevreux suggested the popular "Dummies Guide …" series including Bioinformatics for Dummies (http://www.dummies.com/store/product/Bioinformatics-For-Dummies-2nd-Edition.productCd-0470089857.html) although C. Titus Brown takes issue with the name of these books suggesting that "the culture of "I'm too stupid" inhibits learning". Good point. They look rubbish on your bookshelf too.
Pete @drosophilic suggested enrolling in local training courses, and a list is kept maintained by Stephen Turner at http://stephenturner.us/p/edu. I also note the newly launched iAnn Events platform (http://iann.pro/iannviewer).
Another useful resource is BioStars, see the thread Advice for newcomers to the bioioinformatics field [Pierre Lindenbaum]
C. Titus Brown has a workshop with online course materials for next-generation sequence analysis.
Alan McNally suggested it is possible for a newbie to learn bioinformatics successfully, citing himself as a case study: "I was the requester 4 years ago. Was told to switch to linux and start reading user guides. Haven't looked back".
And finally, Aylwyn Scally remarks that "the first thing I tell them is close MS Excel".
Thanks to all those who took part in the discussion!
Do you have anything to add?
Update: 31st July 2013 - added some links to the Homolog.us blog.
02 May 2013
Bowtie2 defaults to a minimum and maximum insert size of 0 and 500.
Obviously in many cases these defaults are inappropriate and
should be increased.
What is unexpected, for me at least, is the effect these defaults
have on insert size calculation. It seems that this value
actually affects the placement of reads, and therefore
also the calculation of insert sizes.
Perhaps best illustrated with an example, visualised with
Qualimap, with -X 500
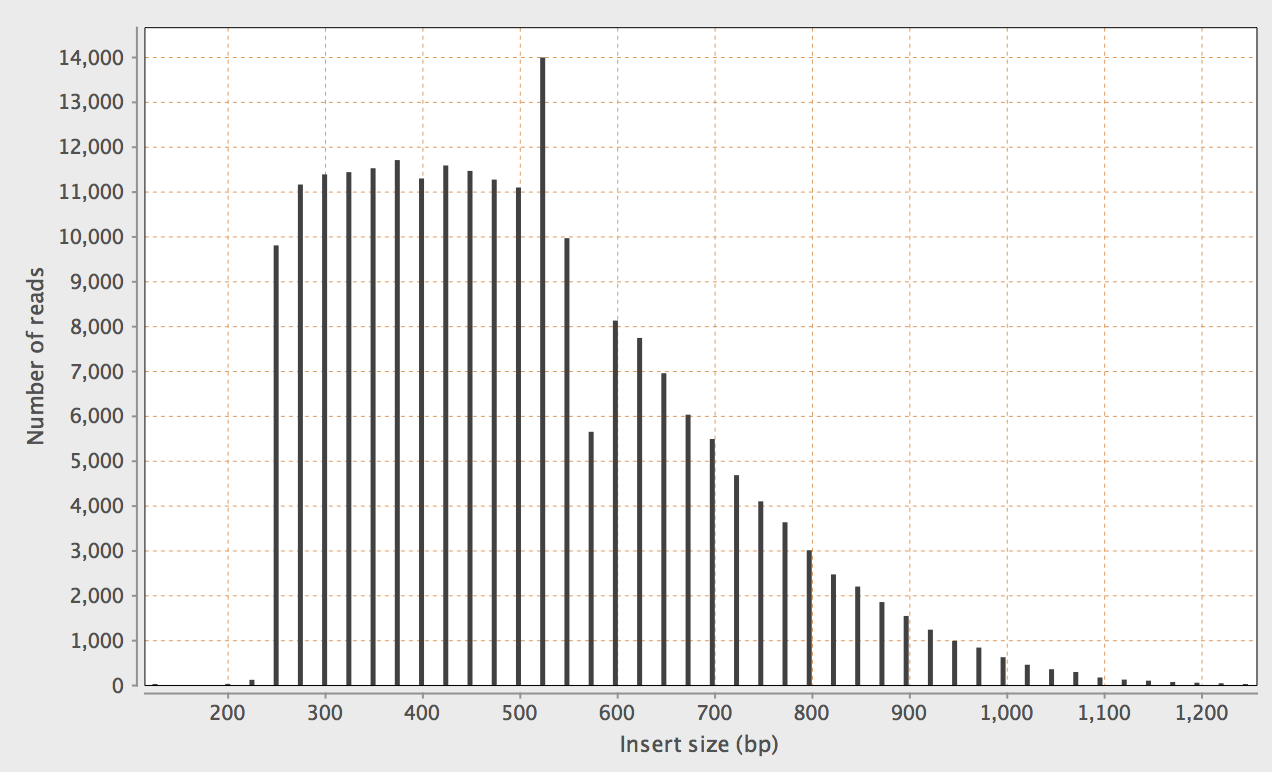
This results in quite an alarming peak. There are
two positions which are significantly enriched at 547-548bp:
samtools view bamfile | cut -f 9 | sort -n | uniq -c
..
421 544
596 545
721 546
1654 547
3050 548
190 549
187 550
212 551
..
Re-running Bowtie2 with -X 1000:
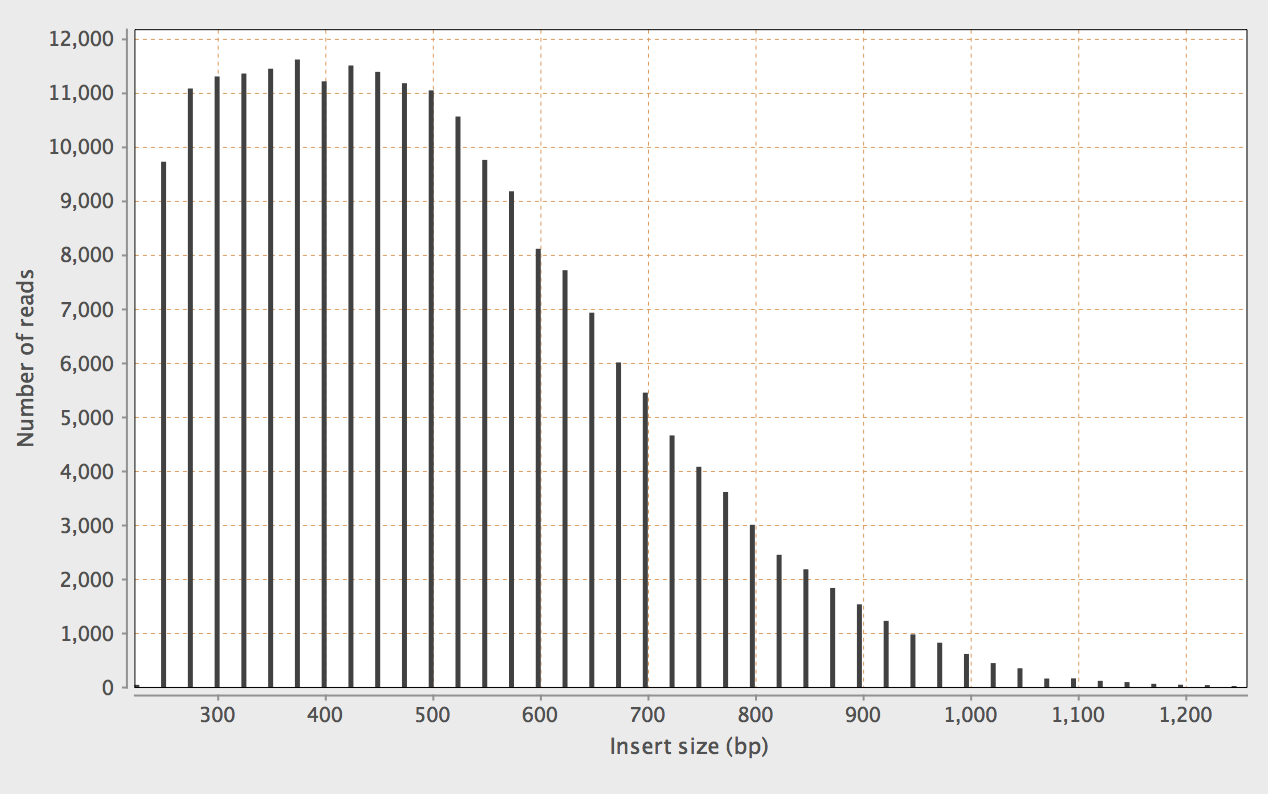
Shows a more appropriate distribution.
I assume this is because Bowtie2 tries to optimise the
choice of alignment to favour those closer to the maximum
insert size? The manual states:
The maximum fragment length for valid paired-end alignments. E.g. if -X 100 is specified and a paired-end alignment consists of two 20-bp alignments in the proper orientation with a 60-bp gap between them, that alignment is considered valid (as long as -I is also satisfied). A 61-bp gap would not be valid in that case. If trimming options -3 or -5 are also used, the -X constraint is applied with respect to the untrimmed mates, not the trimmed mates.
I assume this relates also to the default setting for concordant alignments.
By default, bowtie2 looks for discordant alignments if it cannot find any concordant alignments. A discordant alignment is an alignment where both mates align uniquely, but that does not satisfy the paired-end constraints (–fr/–rf/–ff, -I, -X).
Interestingly this peak position seems to be independent of the species
used. Therefore I assume it relates more to the read length, in this case 2x250.
I tried it with Bowtie2 2.0.2 and 2.1.0.
It might be reasonable to change these defaults now that 2x250 reads are common-place.
Thanks to Josh Quick for figuring out the reason for this.
