Q. How did they sample their environment? How many samples did they look at?
Q. How does this environment compare taxonomically with the human gut? Is it more or less diverse? Are the set of organisms present similar or different?
Q. How does this environment compare functionally with the human gut? Can you explain these findings in the context of the environment?
HOST_SUBJECT_ID - the person each keyboard belongs to
Hint: M1, M2 and M9 are the three participants referred to in the paper.
Q: What are the most abundant taxa?
Q: Check the PCA plots, do samples cluster by key, or by subject (hint: HOST_SUBJECT_ID, )
Q: Go back to the taxa barplots, can you figure out which taxa are driving the variation producing grouping?
Q: Which of these taxa are part of the normal skin microbiome? Are any out of plcae? Where might they come from?
Q: Do you think this technique will really be usable for forensics? What are the challenges? What other techniques might work better for studying the microbiome?
Q: Now, read the paper in more detail and prepare a short summary to present the context for the study, the methods employed and the results found.
Fields of importance: Floor, Level, SURFACE, BUILDING
Q: What surfaces have the greatest amount of diversity? Is this expected?
Q: What do the profiles of stool, etc. look like?
Q: Are there any natural looking clusters in the data?
Q: Which sources of samples are most similar to others?
Q: Is there any clustering between different floors of the building?
Q: Compare the weighted vs unweighted Unifrac results, do the clusters look more natural in one or the toher?
Q: Which surfaces have the most diversity? Least?
Q: Now, read the paper in more detail and prepare a short summary to present to the whole group. Consider: the context for the study, the methods that were employed and the results found. What did you think? What are the limitations of the study?
Q: Is there any evidence of a gradient? (Key: use SampleID and turn gradient colours on)
Q: How do the taxa change over time?
Q: Which infant samples do the maternal stool most look like?
Q: Is the colour of stools associated with their bacterial diversity?
Q: Now, read the paper in more detail and prepare a short summary to present to the whole group. Consider: the context for the study, the methods that were employed and the results found. What did you think? What are the limitations of the study?
“The past is a foreign country” – well, that’s how I feel about January 2016 looking back today. Definitely some things happened in January, but can’t remember them. So I’m using Twitter Analytics to remind me.
Oh! This was the month that #researchparasites came out, to the horror and amusement of the genomics field:
The logical fallacy of #researchparasites: expert data gatherers are highly unlikely to be the best people to analyse their own data.
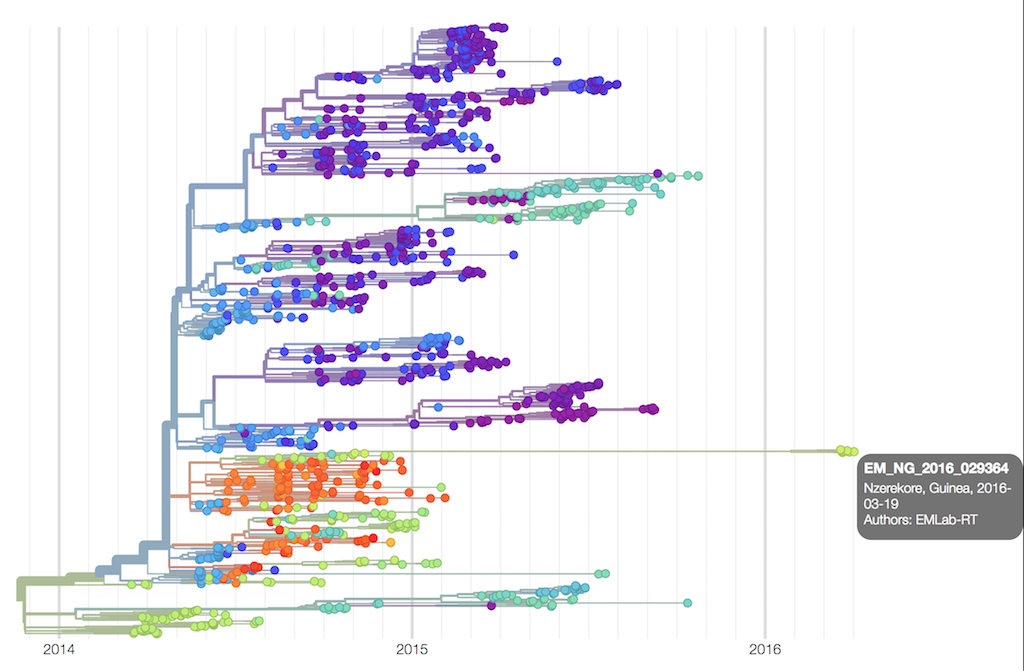
Phylogenetic analysis showed that the new cases were very closely related to an Ebola genome sequenced 500 days previously, as can be seen from this NextStrain tree.
Independently, the epidemiologists identified a survivor who had been infected some 500 days previously, the very same individual.
This was a remarkable demonstration of the power of genomics, working in synergy with the epidemiologists on the ground.
Around the same time, we learnt we had been funded by MRC/Wellcome Trust/Newton to receive funds as part of the emergency response to Zika. Remarkably, the outcome was known just a few weeks after submitting the application, and we had the money just days after that. If only all grant funding could be like this …
We launched the ZiBRA project, a road trip around North-East Brazil to investigate the genomic epidemiology of Zika cases in this region, the most heavily hit by cases of microcephaly in newborns..
We hit the road for the ZiBRA project and started generating Zika genomes working in collaboration with the local public health laboratories. Lots of diaries and blog posts are on the Zibra project website if you want to read more about this trip.
We made lots of new lifelong friends in Brazil, and we didn’t die even though our bus caught on fire at one point, although we hit some technical obstacles with sequencing very very low abundance samples.
The year seemed to be going pretty well. Until the Brexit vote …
Going to bed feeling quite relaxed that the outcome will be fairly strong remain vote tomorrow. Don't make a mockery of this tweet overnight
So far over 150 research groups in the UK have signed up for our virtual machine infrastructure which runs across three sites (Birmingham, Warwick and Cardiff) with Swansea to launch in 2017. Particular props to Radoslaw Poplawski, Tom Connor, Andy Smith, Marius Bakke and Matt Bull on the technical side who helped get this launched - just in time!
August
We sweated over the Zika sequencing protocol and eventually by the end of summer Josh Quick nailed something that worked well on samples with very low viral copy numbers.
In August we just about had time to fit in a week in Cornwall to teach Porecamp with Konrad and his crew.
Pablo, Emily, Jennie and Andy really got MicrobesNG motoring (over 7500 genomes sequenced with a median wait time of 6 weeks!), with insert sizes bolstered with a nice new Nextera XT protocol.
Zika genomes were coming out thick and fast thanks to the new protocol, our Brazilian collaborators, Sarah Hill and Alli Black. Josh’s third trip in 2017. A picture of Zika diversity was now starting to be built (beautifully visualised by Trevor and Richard’s wonderful Nextstrain site - vote for them to win the Open Science prize!).
Well done @scalene, @alliblk and Sarah Hill - smashing out the Zika genomes in Salvador, Brazil with @zibraproject since Friday
All the depressing news on Twitter got too much for me, and I took a break for 3 weeks. After 24 hours of extreme withdrawal symptoms, it was actually quite nice to do something else with my time, like imagining what people were saying on Twitter.
Relentless Brexit and Trump news is turning Twitter into an ordeal rather than a pleasure these days. (I realise this isn't helping).
A relaxed end to the year – we did a bit of Beach (well, Leith) sequencing with Andrew Rambaut and Tom Little in Edinburgh, look out for more BeachSeq action early in 2017..
And we even managed to release data from a 30x human genome on MinION working collaboratively on the sequencing with Nottingham, UCSC, UBC and Norwich, a mere 39 flowcells for that (assembly N50 - 3Mb!):
Finally in December, we heard the great news that the Ebola ca suffit! trial reported 100% efficacy for the Ebola vaccine. Well done Stephan, Miles, Sophie and all the others who worked on this.
A few changes in 2017
The MicrobesNG team sees a change in 2017 - we are sad to say goodbye to Andy Smith - our database programmer on the MicrobesNG project. He’s done an amazing job building the MicrobesNG website, our LIMS, the CLIMB Bryn site, and even had time to help out with the Zibra Project database and the Primal Scheme site. We cannot be too annoyed that he only spent a year with us – he’s had the once in a lifetime opportunity to become a trainee pilot with Aer Lingus, a lifetime dream for Andy.
There have been some changes in Birmingham too – it’s been really nice to have Alan McNally join the IMI as a new Senior Lecturer. And we are really excited that Willem van Schaik is joining the IMI later in April, Brexit be damned!
Politically we are in uncharted territory, so we enter 2017 with some trepidation about what is to happen to the scientific environment, but we also hope that the awesome wins out.
Happy New Year to all friends and collaborators from the Loman Lab!
The nanoporati are currently thrilling to a
bevy of new announcements from Oxford Nanopore Technologies (ONT). More information over at the “wafer-thin update” and insightful commentary from Keith Robison on his blog.
But amongst the noise and excitement of future products, there are three important updates we are focusing on right now:
the release of the 5-10 minute 1D rapid prep (Mu transposase based)
coupled with the new R9 (now R9.4) chemistry that produces usable and high accuracy 1D reads (both discussed in this previous post)
and, new updates to the pore, membrane, motor and loading protocol which suggest 5-10Gb output may now be achievable.
We just received our first R9.4 double-speed (450 b/s) kits and so we will see how it looks soon, but as of now, we are able to get up to 3Gb of output on the vanilla R9.
The significance for our work: we can now start to consider using MinION for metagenomic sequencing (previously we have restricted our ambitions to sequencing individual viruses and bacterial cultures due to relatively low outputs).
Ultimately our research group would like to get to culture-free diagnosis of infectious diseases, with full genomic coverage, as a near-patient assay. There have been a few proof of principle papers here including use on Ebola and chikungunya (from Charles Chiu) and on bacterial urinary tract infections (from Justin O’Grady).
However, for portable metagenomics sequencing to really become a viable prospect, the sample needs to be rapidly prepared at point of collection (from near the patient, in diagnostics, or from water, food, animals, the natural environment, etc.).
In my view, local sample preparation, DNA extraction and local bioinformatics analysis are now the major open issues for portable sequencing.
To illustrate this point, we recently saw the exciting news that the nanopore had been run on the International Space Station - surely a landmark moment in genomics. But yet the sample was still not prepared or DNA extracted in space.
And sadly, you cannot get away without proper DNA extraction. We saw in the past few days the bizarre spectacle of David Eccles and Chris Mason at a conference in Australia attempting to sequence various food samples (coffee, strawberries and cream, etc.) on the nanopore, using the 1D prep. A valiant experiment, but the output was effectively noise due to a lack of pure DNA prep. We experienced similar results when attempting to sequence from a virtually DNA free sample on the beach in Cornwall.
So, DNA extraction remains a fact of life for sequencing.
For single molecule sequencing it’s even trickier: you need high purity, high molecular weight, high concentration DNA to get good results from single molecule sequencers like the nanopore (current input 500ng for the transposon prep).
This should not be problematic for many environmental samples. When dealing with low concentration samples, the easiest way of doing this is via PCR (targeted or untargeted WGA, although fragment length can suffer without significant optimisation).
Solutions for portable sample preparation
Whilst presumably not a big market yet, a few companies have started producing solutions for ‘in-field’ sample preparation and DNA extraction. In the rest of this post I want to explore some of the available options for portable sample preparation and DNA extraction.
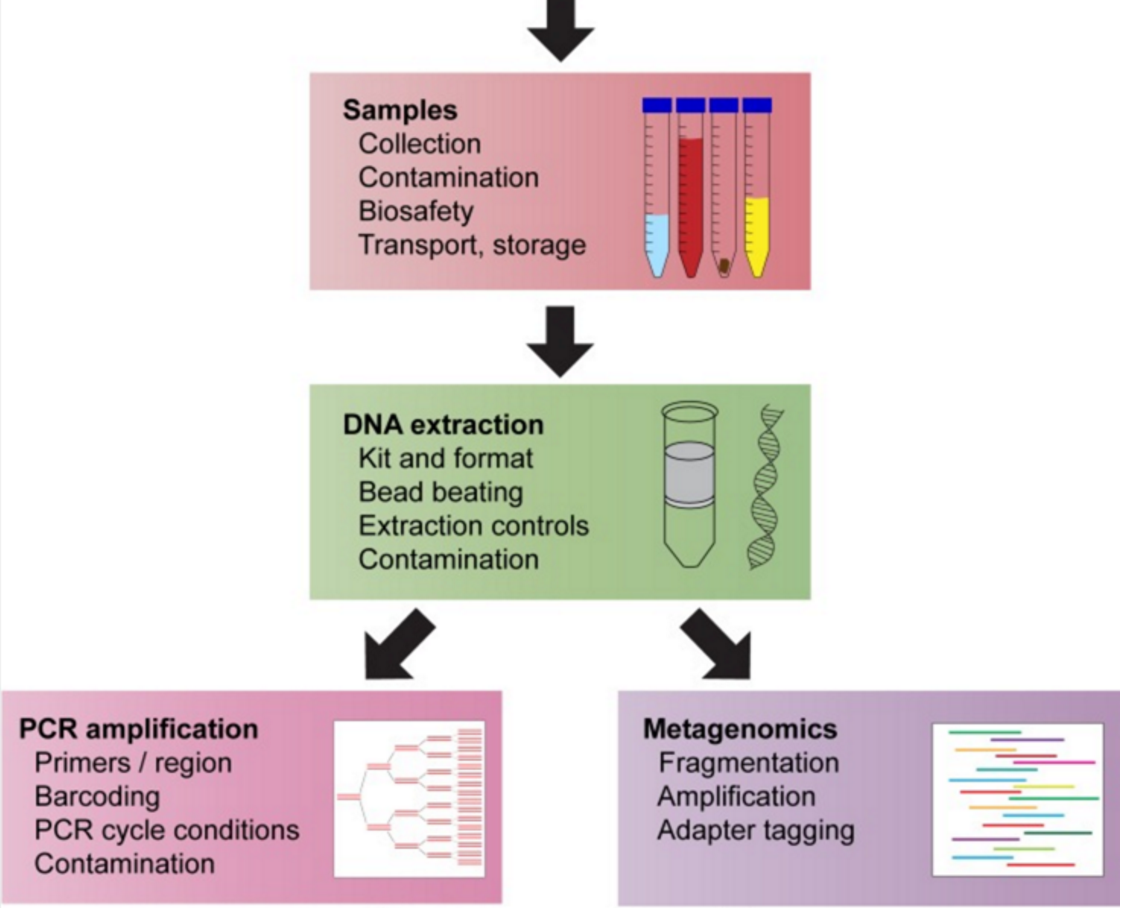
Just as a reminder, the steps for sample preparation are to a) make the sample safe (particularly important e.g. in Ebola) b) homogenise the sample and lyse cells c) extract DNA and then d) make a sequencing library.
Microbiome maven Elizabeth Bik has a very nice review of this in a recent article which is focused around microbiome studies but applies equally to other types of study:
Bead Beating/Tissue Lysis
Many samples, and particularly environmental samples, need homogenisation and cellular disruption before DNA extraction can proceed efficiently. One of the most popular methods is bead beating, which usually requires a benchtop instrument. Luckily, there is a portable, battery-powered method available in the form of the TerraLyzer (we have one). This device available, available from Zymo Research, uses a converted power tool to act as a portable bead beater. It’s a solid bit of kit, but costs about $1000. If that’s too rich for you, Russell Neches has developed a template that can be 3D printed to turn a Craftsman automatic hammer into a portable bead beater.
Here is a video of Russell using the TerraLyzer to extract DNA from cat poo:
DNA extraction
DNA can be extracted very simply from a variety of foods like bananas or strawberries and is method probably familiar to school children; First fruit is blended to break up the tissues, washing up liquid is then added to breakdown the cell membranes before being straining to remove solids. DNA is then precipitated by adding alcohol and spooled off using a toothpick. More detailed instructions here: http://biology.about.com/od/biologylabhowtos/ht/dnafromabanana.htm.
100% ethanol is a problematic substance to ship (it is banned from aircrafts), so it is an open research question about whether a lower proof alcohol that is readily available, e.g. vodka, would be an acceptable substitute. Please fund this important project.
Portable devices
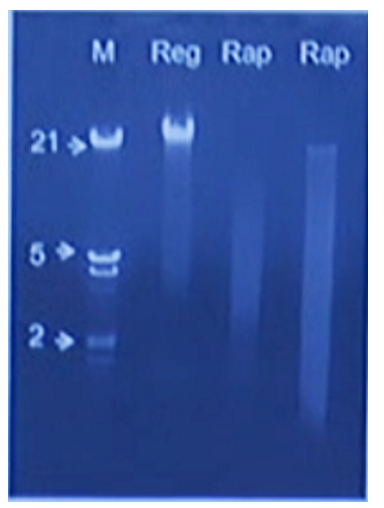
Claire Lonsdale brought along an interesting device to Porecamp in Cornwall called the PureLyse from Claremont Bio. This device combines bead-beating with DNA capture using silica beads which are agitated by a small motor. They have built a small disposable device which combines a syringe and a reusable battery pack. The sample, ideally bacterial culture, is aspirated via the syringe then the motor is turned on for a minute to burst the cells/bind the DNA. Claire presented results at London Calling demonstrating that the DNA extracted is probably suitable for PCR but may be too fragmented for single-molecule sequencing.
Screenshot from Claire’s London Calling talk, showing rapid extraction on the right compared to a regular spin-column extraction on the left.
The announced, but not currently available Zumbador from ONT looks to take this syringe concept further, by including reagents for lysis, purification and potentially library preparation in a single pre-loaded cartridge. This looks appealing but the worry for those of us who deal with a lot of DNA extractions from different organisms is which cell lysis solution is likely to be universally applied to all manner of organisms with quite different cell wall compositions - Gram positives and spore forming bacteria are notoriously tough shells to crack, this may need to be combined with the bead beating step above.
Zymo also offer the Xpedition kit range, which are designed with field work in mind and contain a stabilisation solution which will preserve your DNA (after bead beating with the TeraLyzer) for up to a month at room temperature.
However, you can also use traditional column-based extraction method in the field, if you have a:
Portable centrifuge
From my research, microcentrifuges are nearly all mains powered, which limits their utility in the field.
A homebrew solution is simply to modify a cordless drill with a 3D printed centrifuge adaptor, one example being the DremelFuge, that offers up to 52,000g/rcf acceleration.
However one should be extremely careful here because a flying, solid object at these rotations could cause serious harm, please take appropriate safety precautions if you are thinking of using this solution. Disclaimer! More generally, if in doubt about any safety aspects of field sample preparation, please first get in contact with your local safety officer for advice.
An alternative is to adapt a regular lab microcentrifuge that can take DC input, as that means they can be easily powered from a Lithium-Ion battery pack.
Portable PCR Thermocyclers
The MiniPCR is a fantastic (we’ve got one) biohacker/kickstarter product which costs £500 from Cambio in the UK. It is programmed via a laptop or phone but then must be plugged into a mains adapter or battery pack to start the program. We bought a LiPo powerbank off Amazon for £70 which can provide the 19V, 3.7A power requirement. They also produce a small electrophoresis and visualisation system to go with it.
An alternative is the Bento Lab from Bento Bio. This device caught many people’s attention with its Fisher-Price toy looks and intriguing functionality - it is a PCR thermocycler, gel visualiser block and minifuge all in one! Although mains powered, it should draw sufficiently little power that it could be powered via a car battery or possibly a Lithium pack. We had the pleasure of seeing a prototype box and it kicks ass - the only problem at the moment is that it’s still not available to buy. I hope it will ship soon and we’ll be first in the queue to test it out.
## Portable Liquid Handler
Pipetting is only accurate at relatively large volumes (>1 ul) which both increases reagent costs and can be a major source of errors with multi-step protocols. The Voltrax is an interesting device that was announced by ONT at London Calling 2015 and has not yet been seen in the wild, although the access programme was recently announced. The basic principle is the movement of ultra low liquid volumes around a matrix through an applied electrical current - a process called electrowetting. The appeal of such a process is that complex pipetting and mixing steps could be automated (apparently via a scriptable Python interface).
There may well be more that I have not mentioned … feel free to drop your suggestions in the comments box below!
Conflict of interests
I have received an honorarium to speak at an Oxford Nanopore meeting, and travel and accommodation to attend London Calling 2015 and 2016. I have ongoing research collaborations with ONT although I am not financially compensated for this and hold no stocks, shares or options. ONT have supplied free-of-charge reagents as part of the MinION Access Programme and also generously supported our infectious disease surveillance projects with reagents. Cambio sent us some free reagents to go with the MiniPCR instrument we purchased.
Credits
Thanks to Josh Quick for contributing to this post, and to Matt Loose and John Tyson for reading a draft version.
A long promised addition to the nanopore sequencing repertoire is the rapid sequencing kit. This kit significantly reduces the effort required to make a sequencing library - down from 2-3 hours to a few minutes. We’ve actually played with this kit several times before, once very early on in the MAP (I think using R7 chemistry as long ago as July 2014). More recently, Matt Loose and I tried it out in a hotel room before a famous genomics conference in February of this year. We can both vouch for how easy it is to use - no specialist equipment is required other than pipettes and a source of heat to neutralise the transposase after a short incubation at room temperature. The recommended starting DNA input is 500ng. In our hotel room we used a freshly brewed cup of coffee which provided the required 70 degrees.
However, until recently this kit was really mainly a curiosity rather than a serious proposition because it only produces so-called “1D” data. To remind you, 1D data is when only the template strand of the double-stranded molecule is read. With the 1D kit because there is no hairpin ligation the complement strand does not pass through the pore.
And for R7.3 data this was a significant drawback: sequence accuracy on the template strand is in the low 70s, accuracy-wise, which makes basic tasks like de novo assembly and variant calling computationally very difficult (although probably not impossible, and assemblers like Canu can cope, with a bit of tweaking). It also makes polishing extremely slow.
The release a few months back of the R9 chemistry has changed the game – it’s a game-changer! – and suddenly made 1D reads very usable. This is ascribed to the more discriminatory read head of the CsgG pore employed, where fewer nucleotides in the pore abrogate the flow of ions across the membrane. The spread of electrical current levels is about twice as wide as seen in R7. However it is hard to know exactly how much of the improved accuracy is caused by the pore as this coincided with the introduction of a new style of basecaller that employs ‘deep learning’ (technically a recurrent neural network) rather than the Hidden Markov Model of before. A third change is the introduction of ‘fast mode’, currently running at 250 bases / second, or four times the translocation speed employed with the R7 chemistry. Because all these changes were introduced at once, it is hard to know the relative contribution of each. However, our early access experiences with R7.3 demonstrated that ‘fast mode’ did not seem to have a significant detrimental effect on quality. In fact, the theory is it may improve handling of long homopolymeric tracts by introducing more signal into the ‘dwell’ times.
Other changes: Notably, the sequencing files now record raw current sample data (at 5kHz) by default, and the previous process of linearising the signal into ‘events’ is now performed by the cloud base caller Metrichor rather than MinKNOW on the laptop. Excitingly there are now three local basecallers available - one is built into MinKNOW 1.0.0 (the next release). There is also a separate download called nanonet (available to MAPpers). We tried out nanonet during the ZiBRA bus trip and it worked well, albeit it could not quite keep up with data generation on a standard laptop. Jared Simpson and Matei David also have an open source basecaller called nanocall.
We’ve done two runs of this protocol. The first was on a flowcell that was delivered, erroneously frozen for 36 hours at -10 degrees in our Stores, and then left at room temperature for a week or so (we’d assumed it was completely knackered). We thought we’d just try it out for fun and to our surprise it actually generated a decent yield of data, around 600mb. Data here is from a second flowcell that was correctly stored at fridge temperature.
The final new thing here is that this is a SpotON flowcell; which means the total volume loaded onto the flowcell is halved, and you in fact ‘drip, drip’ the library straight onto the flowcell surface via a small hole that is protected by a plastic clip. What difference this makes to performance is currently unknown:
The results from the better flowcell are presented here with links to data at the bottom:
E. coli stats
stats
Type
Total Reads
Base Pairs
Mean
Median
Min
Max
N25
N50
N75
pass:template
164472
1.48Gb
9009
5944
117
131969
25244
14891
8074
fail:template
74465
467Mb
6271
3544
5
328471
21903
12033
6047
This is the highest yielding flowcell we’ve ever had, with just shy of 2Gb of base called sequence, and 1.48Gb in the pass bin. Over 99% of the reads map to the reference, meaning the goodput is equivalent to the output.
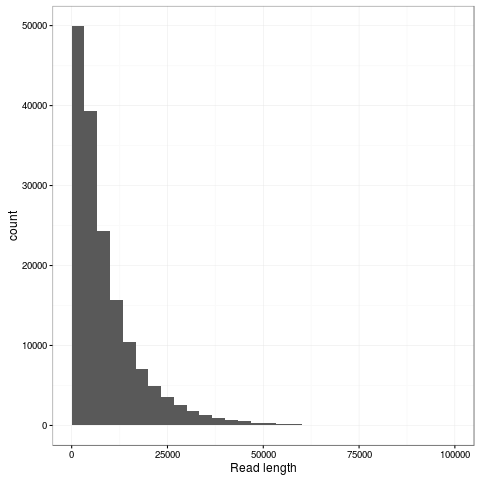
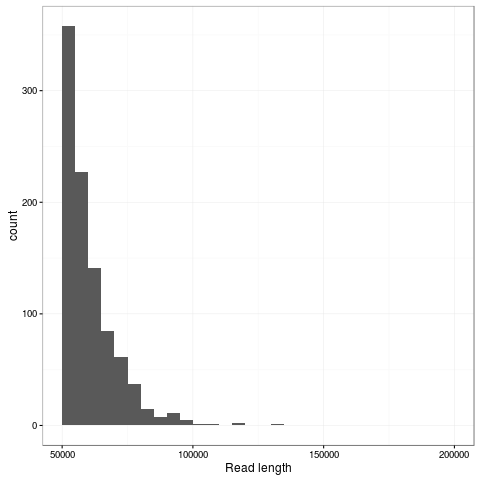
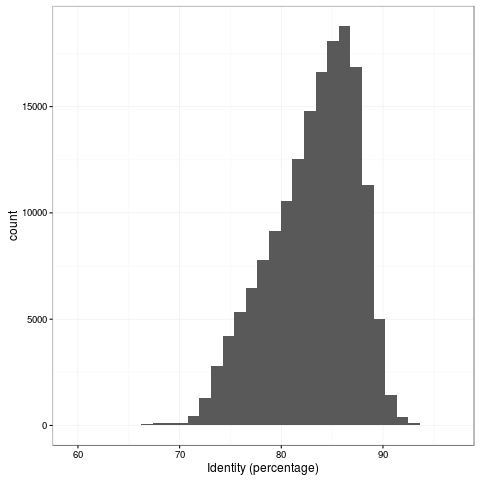
The transpososome method gives a very different size distribution to the Gaussian distribution expected with the traditional Covaris G-tube fragmentation. There are more shorter reads, but the N50 is improved to nearly 15kb (from around 8kb). The maximum length read in this dataset is 131kb and aligns completely to the reference genome at 85% identity.
Zooming into this plot it is obvious there are plenty of super long reads - 953 of the passing reads are greater than 50kb comprising 57.5Mb of sequence.
Gratifyingly the data gives a single contig assembly with miniasm and Canu without any custom parameterisation. We’ll pass it over to Jared to see what kind of consensus accuracy he can get out of nanopolish which now has alpha support for R9 data.
The 1D accuracy is a quantum leap from previous pores, with mean read accuracy at 83%.
We’ll do more analysis on this dataset and hope to write it up as a manuscript in future, but are releasing the dataset for the community to play with.
I have posted up the IPython notebook detailing the commands to reproduce this analysis.
Credits
Josh Quick did the laboratory work and sequencing. We are grateful to John Tyson for supplying his tuning scripts for the 1D R9 run.
Conflict of interests
I have received an honorarium to speak at an Oxford Nanopore meeting, and travel and accommodation to attend London Calling 2015 and 2016. I have ongoing research collaborations with ONT although I am not financially compensated for this and hold no stocks, shares or options. ONT have supplied free-of-charge reagents as part of the MinION Access Programme and also generously supported our infectious disease surveillance projects with reagents.